Pipes y filtros
Última actualización: 2023-04-24 | Mejora esta página
Hoja de ruta
Preguntas
- ¿Cómo puedo combinar comandos existentes para hacer cosas nuevas?
Objetivos
- Redireccionar la salida de un comando a un archivo.
- Procesar un archivo en lugar de la entrada de teclado mediante la redirección.
- Construir pipelines de comandos con dos o más etapas.
- Explicar lo que normalmente sucede si un programa o un pipeline no recibe ninguna entrada para procesar.
- Explicar las filosofía de Unix de ‘pequeñas piezas, libremente unidas’.
Ahora que ya sabemos algunos comandos básicos, podemos ver finalmente
la característica más poderosa de la terminal: la facilidad con la que
nos permite combinar los programas existentes de nuevas maneras.
Comenzaremos con un directorio llamado molecules que
contiene seis archivos que describen algunas moléculas orgánicas
simples. La extensión .pdb indica que estos archivos están
en formato Protein Data Bank, un formato de texto simple que especifica
el tipo y la posición de cada átomo en la molécula.
SALIDA
cubane.pdb ethane.pdb methane.pdb
octane.pdb pentane.pdb propane.pdbEntra a ese directorio usando el comando cd y ejecuta el
comando wc *.pdb. wc es el comando “word
count”: cuenta el número de líneas, palabras y caracteres de un archivo.
El * en *.pdb coincide con cero o más
caracteres, por lo que la terminal convierte *.pdb en una
lista de todos los archivos .pdb en el directorio
actual:
SALIDA
20 156 1158 cubane.pdb
12 84 622 ethane.pdb
9 57 422 methane.pdb
30 246 1828 octane.pdb
21 165 1226 pentane.pdb
15 111 825 propane.pdb
107 819 6081 totalCaracteres especiales
* Es un caracter especial o wild card.
Corresponde a cero o más caracteres, así que *.pdb coincide
conethane.pdb, propane.pdb, y cada archivo que
termina con ‘.pdb’. Por otro lado, p*.pdb sólo coincide con
pentane.pdb ypropane.pdb, porque la ‘p’ al
inicio hace coincidir los nombres de archivos que comienzan con la letra
‘p’.
? también es un caracter especial, pero sólo coincide
con un solo carácter. Esto significa que p?.pdb podría
coincidir con pi.pdb op5.pdb (si existieran en
el directorio molecules), pero nopropane.pdb.
Podemos usar cualquier número de caracteres especiales a la vez: por
ejemplo, p*.p?* coincide con cualquier cosa que comience
con una ‘p’ y termine con ‘.’, ‘p’, y al menos un carácter más (ya que
? tiene que coincidir con un carácter, y el final
* puede coincidir con cualquier número de caracteres). Por
lo tanto, p*.p?* coincidirá con
preferred.practice, e incluso p.pi (dado que
el primer * no coincide con ningún carácter), pero no
quality.practice (ya que no inicia con ‘p’) o
preferred.p (porque no hay al menos un carácter después de
‘.p’).
Cuando la terminal reconoce un caracter especial lo expande para
crear una lista de nombres de archivo coincidentes antes de
ejecutar el comando seleccionado. Como excepción, si una expresión con
caracteres especiales no coincide con algún archivo, la terminal pasará
la expresión como un parámetro al comando tal y como está escrita. Por
ejemplo, ejecutar ls *.pdf en el
directoriomolecules (que contiene sólo los archivos con
nombres que terminan con .pdb) da como resultado un mensaje
de error indicando que no hay ningún archivo llamado
* .pdf. Sin embargo, generalmente comandos como
wc yls ven las listas de nombres de archivo
que coinciden con estas expresiones, pero no los comodines por si solos.
Es la terminal, no los otros programas, la que se ocupa de expandir los
caracteres especiales, este es otro ejemplo de diseño ortogonal.
Usando caracteres especiales
En el directorio molecules, ¿qué variación del
comandols producirá esta salida?
ethane.pdb methane.pdb
ls *t*ane.pdbls *t?ne.*ls *t??ne.pdbls ethane.*
La solución es 3.
muestra todos los archivos que contienen cualquier número o combinación de caracteres seguidos de la letra
t, otro caracter único, y terminan conane.pdb. Esto incluyeoctane.pdbypentane.pdb.muestra todos los archivos que contienen cualquier número o combinación de caracteres,
t, otro caracter,ne., seguido de otro número o combinación de caracteres. Esto regresaríaoctane.pdbypentane.pdbpero nad que termine conthane.pdb.soluciona el problema de la opción 3 agregando dos caracteres entre
tyme. Esta es la opción correcta.sólo muestra archivos que comienzan con
ethane..
Si ejecutamos wc -l en lugar dewc, la
salida sólo muestra el número de líneas por archivo:
SALIDA
20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 totalTambién podemos usar -w para obtener sólo el número de
palabras, o -c para obtener sólo el número de
caracteres.
¿Cuál de estos archivos es el más corto? Es una pregunta fácil de responder cuando sólo hay seis archivos, pero ¿y si hubiera 6,000? Nuestro primer paso hacia una solución es ejecutar el comando:
El símbolo “mayor que”, >, le dice a la terminal que
redireccione la salida del comando a un archivo en
lugar de imprimirlo en la pantalla. (Es por eso que no hay salida de
pantalla: en vez de mostrarlo, todo lo que wc imprime se ha
enviado al archivo lengths.txt.) Si no existe el archivo,
la terminal lo creará. Si el archivo existe, será sobrescrito
silenciosamente, lo que puede provocar pérdida de datos y, por lo tanto,
requiere cierta precaución. ls lengths.txt confirma que el
archivo existe:
SALIDA
lengths.txtAhora podemos enviar el contenido de lengths.txt a la
pantalla usando cat lengths.txt. cat significa
“concatenate” (concatenar): imprime el contenido de los archivos uno
tras otro. En este caso sólo hay un archivo, así que cat
sólo nos muestra lo que éste contiene:
SALIDA
20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 totalSalida Página por página
Continuaremos usando cat en esta lección, por
conveniencia y consistencia, pero tiene la desventaja de que siempre
vuelca todo el archivo en la pantalla. En la práctica es más útil el
comando less, que se utiliza como
$ less lengths.txt. Este comando muestra sólo el contenido
del archivo que cabe en una pantalla y luego se detiene. Puedes avanzar
a la siguiente pantalla presionando la barra espaciadora, o retroceder
presionando b. Para salir, pulsa q.
Ahora utilizemos el comando sort para ordenar el
contenido. También usaremos el indicador -n para
especificar que el tipo de orden que requerimos es numérico en lugar de
alfabético. Esto no cambia el archivo; sólo despliega el
resultado ordenado en la pantalla:
SALIDA
9 methane.pdb
12 ethane.pdb
15 propane.pdb
20 cubane.pdb
21 pentane.pdb
30 octane.pdb
107 totalPodemos poner la lista de líneas ordenada en otro archivo temporal
llamado sorted-lengths.txt poniendo
> sorted-lengths.txt después del comando, así como
usamos > lengths.txt para poner la salida de
wc en lengths.txt. Una vez que hayamos hecho
eso, podemos ejecutar otro comando llamado head para
obtener las primeras líneas de sorted-lengths.txt:
SALIDA
9 methane.pdbEl parámetro -n 1 con head indica que sólo
queremos la primera línea del archivo; -n 20 conseguirá las
primeras 20, y así sucesivamente. Dado que
sorted-lengths.txt contiene las longitudes de nuestros
archivos ordenados de menor a mayor, la salida de head debe
ser el archivo con menos líneas.
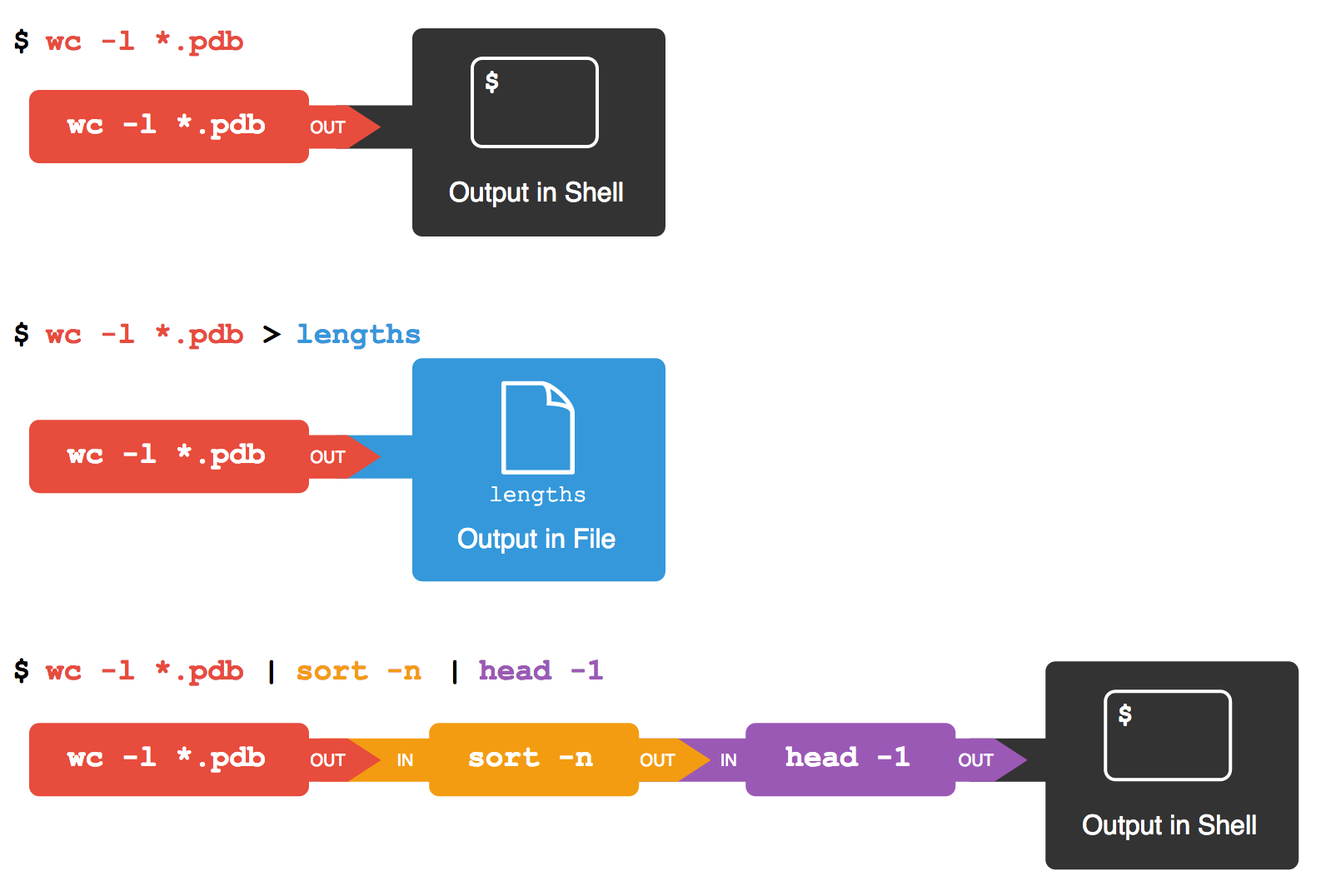
Si crees que es confuso, no estás solo: incluso una vez que entiendas
lo que wc,sort y head hacen,
todos esos archivos intermedios hacen difícil seguir el hilo de lo que
está pasando. Podemos hacerlo más fácil de entender ejecutando
sort y head juntos:
SALIDA
9 methane.pdbLa barra vertical, |, entre los dos comandos se denomina
pipe (pronunciado paip). El pipe le
dice a la terminal que queremos usar la salida del comando a la
izquierda como entrada al comando de la derecha. La computadora puede
crear un archivo temporal si es necesario, copiar datos de un programa a
otro en la memoria, o cualquier otra cosa que sea necesaria; no es
necesario que lo entendamos para hacerlo funcionar.
Nada nos impide encadenar pipes consecutivamente.
Por ejemplo, puedes enviar la salida de wc directamente a
sort, y luego la salida resultante a head.
Así, primero usamos un pipe para enviar la salida de
wc a sort:
SALIDA
9 methane.pdb
12 ethane.pdb
15 propane.pdb
20 cubane.pdb
21 pentane.pdb
30 octane.pdb
107 totalY ahora enviamos la salida de este pipe, a través de
otro pipe, a head, para que el
pipeline completo se convierta en:
SALIDA
9 methane.pdbEsto es exactamente como un matemático anidando funciones como
log(3x) Y diciendo “el logaritmo de tres veces x”. En
nuestro caso, el cálculo es “cabeza de la lista ordenada del número de
líneas de *.pdb”.
Esto es lo que realmente sucede detrás de la terminal cuando creamos un pipe. Cuando una computadora ejecuta un programa (cualquier programa) crea un proceso en memoria para almacenar el software del programa y su estado actual. Cada proceso tiene un canal de entrada llamado entrada estándar. (Para este punto, puede sorprenderte que el nombre es tan memorable, pero no te preocupes, la mayoría de los programadores de Unix lo llaman “stdin”). Cada proceso también tiene un canal de salida predeterminado llamado salida estándar (o “stdout”). Un tercer canal de salida llamado error estándar (stderr) también existe. Este canal suele utilizarse para mensajes de error o de diagnóstico y permite al usuario canalizar la salida de un programa a otro mientras sigue recibiendo mensajes de error en el terminal.
La terminal es realmente otro programa. Bajo circunstancias normales, lo que ingresemos en el teclado se envía a la entrada estándar de la terminal, y lo que produce en la salida estándar se muestra en nuestra pantalla. Cuando le decimos a la terminal que ejecute un programa, ésta crea un nuevo proceso y envía temporalmente lo que tecleamos en nuestro teclado a la entrada estándar de ese proceso, y lo que el proceso envía a la salida estándar, la terminal lo envía a la pantalla.
Esto es lo que ocurre cuando ejecutamos
wc -l *.pdb > lengths.txt. La terminal comienza
diciéndole a la computadora que cree un nuevo proceso para ejecutar el
programa wc. Como hemos proporcionado algunos nombres de
archivo como parámetros, wc lee estos en vez de la entrada
estándar. Y puesto que hemos utilizado > para redirigir
la salida a un archivo, la terminal conecta la salida estándar del
proceso a ese archivo.
Si ejecutamos wc -l *.pdb | sort -n en su lugar, la
terminal crea dos procesos (uno para cada proceso en el
pipe) de modo que wc ysort
funcionan simultáneamente. La salida estándar de wc es
alimentada directamente a la entrada estándar de sort; ya
que no hay redirección con >, la salida de
sort va a la pantalla. Y si ejecutamos
wc -l * .pdb | sort -n | head -n 1, obtenemos tres procesos
con datos que fluyen de los archivos, a través de wc a
sort, de sort ahead y finalmente
a la pantalla.
Esta sencilla idea es la razón por la cual Unix ha tenido tanto
éxito. En lugar de crear enormes programas que tratan de hacer muchas
cosas diferentes, los programadores de Unix se centran en crear muchas
herramientas simples que hacen bien su trabajo y son capaces de cooperar
entre sí. Este modelo de programación se llama “pipes y
filtros”. Ya hemos visto pipes; un
filtro es un programa como wc
osort que transforma una entrada en una salida. Casi todas
las herramientas estándar de Unix pueden funcionar de esta manera: a
menos que se les indique lo contrario, leen de entrada estándar, hacen
algo con lo que han leído, y escriben en la salida estándar.
La clave es que cualquier programa que lea líneas de texto de entrada estándar y escriba líneas de texto en la salida estándar puede combinarse con cualquier otro programa que se comporte de esta manera también. Puedes y debes escribir tus programas de esta manera para que tú y otras personas puedan poner esos programas en pipes y así multiplicar su poder.
Redireccionamiento de entrada
Además de usar > para redirigir la salida de un
programa, podemos usar < para redirigir su entrada, por
ejemplo, para leer un archivo en lugar de la entrada estándar. En lugar
de escribir wc ammonia.pdb, podríamos escribir
wc < ammonia.pdb. En el primer caso, wc
obtiene un parámetro de línea de comandos diciéndole qué archivo abrir.
En el segundo, wc no tiene ningun parámetro de la línea de
comandos, por lo que se lee desde la entrada estándar, pero hemos
indicado al shell que envíe el contenido de ammonia.pdb a
la entrada estándar de wc, por lo que el resultado de ambos
comandos es el mismo.
Pipeline de Nelle: Comprobación de archivos
Nelle ha procesado sus muestras en su máquina de ensayo, generando 17
archivos en el directorio north-pacific-gyre/2012-07-03
descrito anteriormente. Como un chequeo rápido, a partir de su
directorio de inicio, Nelle teclea:
La salida son 18 líneas que como estas:
SALIDA
300 NENE01729A.txt
300 NENE01729B.txt
300 NENE01736A.txt
300 NENE01751A.txt
300 NENE01751B.txt
300 NENE01812A.txt
... ...Ahora escribe esto:
SALIDA
240 NENE02018B.txt
300 NENE01729A.txt
300 NENE01729B.txt
300 NENE01736A.txt
300 NENE01751A.txtUps: uno de los archivos tiene 60 líneas menos que los otros. Cuando Nelle vuelve y lo revisa ve que hizo ese ensayo a las 8:00 un lunes por la mañana. Alguien probablemente usó la misma máquina ese fin de semana, y olvidó reiniciarla. Antes de volver a analizar esa muestra, decide comprobar si algunos archivos tienen demasiados datos:
SALIDA
300 NENE02040B.txt
300 NENE02040Z.txt
300 NENE02043A.txt
300 NENE02043B.txt
5040 totalEsas cifras parecen buenas pero, ¿qué es esa ‘Z’ en la antepenúltima línea? todas sus muestras deben estar marcadas con “A” o “B”; por convención, su laboratorio utiliza ‘Z’ para indicar muestras con información que falta. Para encontrar a otros archivos como este, Nelle hace lo siguiente:
SALIDA
NENE01971Z.txt NENE02040Z.txtComo esperaba, cuando comprueba el registro en su computadora
portátil, no hay profundidad registrada para ninguna de esas muestras.
Ya que es demasiado tarde para obtener la información de otra manera,
ella debe excluir esos dos archivos de su análisis. Podría simplemente
borrarlos usando rm, pero en realidad hay algunos análisis
que podría hacer más tarde, donde la profundidad no importa, por lo que,
en vez de borrarlos, sólo tendrá cuidado al seleccionar archivos
utilizando la expresión con caracteres especiales
*[AB].txt. Como siempre, el * coincide con
cualquier número de caracteres; la expresión [AB] coincide
con una ‘A’ o una ‘B’, por lo que coincide con los nombres de todos los
archivos de datos válidos que tiene.
¿Qué hace sort -n?
Si ejecutamos sort en este archivo:
10
2
19
22
6la salida es:
SALIDA
10
19
2
22
6Si ejecutamos sort -n en la misma entrada, obtendremos
esto en su lugar:
SALIDA
2
6
10
19
22Explique por qué -n tiene este efecto.
La opción -n indica un ordenamiento numérico, en lugar
de alfabético.
< se usa para redirigir la entrada a un comando.
En los dos ejemplos, la terminal regresa el número de líneas en la
entrada al comando wc. En el primer ejemplo, la entrada es
el archivo notes.txt y el nombre del archivo se menciona en
la salida del comando wc. En el segundo ejemplo, el
contenido del archivo notes.txt es redireccionado a la
entrada estándar. Es como si hubiéramos introducido el contenido del
archivo a mano. Por esto, el nombre del archivo no es especificado en la
salida, sólo el número de líneas. Inténtalo por tu cuenta.
BASH
$ wc -l
this
is
a test
Ctrl-D # Esto le indica a la terminal que has terminado de teclear la entrada.SALIDA
3¿Qué significa
>>?Hemos visto el uso de
>pero hay un operador similar>>que funciona un poco distinto. Aprenderemos sobre la diferencia de estos comandos impriendo algunas cadenas de caracteres. Podemos usar el comandoechopara imprimir cadenas de caracteres, e.g.:
SALIDA
El comando echo imprime el texto
Ahora prueba los comandos siguientes para revelar la diferencia entre los dos operadores.
¿Cuál es la diferencia entre:
Más información sobre caracteres especiales
Sam tiene un directorio que contiene datos de calibración, otros datos y descripciones de estos datos:
BASH
2015-10-23-calibration.txt
2015-10-23-dataset1.txt
2015-10-23-dataset2.txt
2015-10-23-dataset_overview.txt
2015-10-26-calibration.txt
2015-10-26-dataset1.txt
2015-10-26-dataset2.txt
2015-10-26-dataset_overview.txt
2015-11-23-calibration.txt
2015-11-23-dataset1.txt
2015-11-23-dataset2.txt
2015-11-23-dataset_overview.txtAntes de ir a otro viaje de campo, Sam quiere respaldar sus datos y enviar algunos datasets a su colega Bob. Sam utiliza los siguientes comandos para lograrlo:
BASH
$ cp *dataset* /backup/datasets
$ cp ____calibration____ /backup/calibration
$ cp 2015-____-____ ~/send_to_bob/all_november_files/
$ cp ____ ~/send_to_bob/all_datasets_created_on_a_23rd/Ayuda a Sam rellenando los espacios en blanco.
Uniendo comandos con pipes
En nuestro directorio actual, queremos encontrar los 3 archivos que tienen el menor número de líneas. ¿Cuál de los siguientes comandos funcionaría?
wc -l * > sort -n > head -n 3wc -l * | sort -n | head -n 1-3wc -l * | head -n 3 | sort -nwc -l * | sort -n | head -n 3
La solución es la opción 4. El caracter de pipe
| es usado para alimentar la entrada estándar de un proceso
con la salida estándar del anterior. > es utilizado para
redirigir la salida estándar a un archivo. ¡Inténtalo en el directorio
data-shell/molecules!
¿Por qué uniq sólo elimina
duplicados adyacentes?
El comando uniq elimina las líneas duplicadas adyacentes
de su entrada. Por ejemplo, el archivo
data-shell/salmon.txt contiene:
coho
coho
steelhead
coho
steelhead
steelheadEl comando uniq salmon.txt del directorio
data-shell/data produce:
SALIDA
coho
steelhead
coho
steelhead¿Por qué crees que uniq sólo elimina líneas
adyacentes duplicadas? (Pista: piensa en datasets muy largos.) ¿Qué
otro comando podría combinarse con él en un pipe para
eliminar todas las líneas duplicadas?
Comprensión de la lectura de pipes
Un archivo llamado animals.txt (en el directorio
data-shell/data) contiene los siguientes datos:
2012-11-05,deer
2012-11-05,rabbit
2012-11-05,raccoon
2012-11-06,rabbit
2012-11-06,deer
2012-11-06,fox
2012-11-07,rabbit
2012-11-07,bear¿Qué texto pasa a través de cada uno de los pipes y qué incluye el redireccionamiento final en el siguiente pipeline?
Pista: construye el pipeline agregando un comando a la vez para comprobar tu respuesta.
Construcción de Pipes
Para el archivo animals.txt del ejercicio anterior, el
comando:
produce la siguiente salida:
SALIDA
deer
rabbit
raccoon
rabbit
deer
fox
rabbit
bear¿Qué otro(s) comando(s) podría(n) agregarse a estos en un pipeline para encontrar qué animales contiene el archivo (sin nombres duplicados)?
Eliminación de archivos innecesarios
Suponte que deseas borrar tus archivos de datos procesados y sólo
conservar los archivos sin procesar y el script de
procesamiento para ahorrar espacio en disco. Los archivos sin procesar
terminan en .dat y los archivos procesados terminan en
.txt. ¿Cuál de las siguientes opciones eliminaría todos los
archivos de datos procesados, y nada más que los archivos de
datos procesados?
rm ?.txtrm *.txtrm * .txtrm *.*
- Esto eliminaría archivos con terminación
.txty nombres de un caracter. - Esta es la respuesta correcta.
- La terminal expandiría el
*para coincidir con todo el directorio actual, así que el comando intentaría eliminar todos los archivos, más un archivo adicional llamado.txt. - La terminal expandiría
*.*para coincidir con todos los archivos, independientemente de su terminación. Este comando eliminaría todos los archivos.
Expresiones con Caracteres Especiales
Las expresiones con caracteres especiales pueden llegar a ser muy
complejas, sin embargo a veces es posible escribirlas de forma que
utilicen una sintaxis muy sencilla y sean un poco más elocuentes.
Considera el directorio
data-shell/north-pacific-gyre/2012-07-03 : la expresión
*[AB].txt coincide con todos los archivos que terminan en
A.txt o B.txt. Imagina que ignoras esto.
¿Podrías hacer coincidir el mismo set de archivos con una expresión que incluya caracteres especiales, pero no
[]? Pista: Podrías necesitar más de una expresión.La expresión que encontraste y la expresión usada en la lección coinciden con el mismo grupo de archivos en el ejemplo. ¿Puedes identificar la ligera diferencia en la salida de ambos comandos?
¿Bajo qué circunstancias tu nueva expresión produciría un mensaje de error y la expresión original no?
¿Qué tubería?
El archivo data-shell/data/animals.txt contiene 586
líneas de datos en el siguiente formato:
SALIDA
2012-11-05,deer
2012-11-05,rabbit
2012-11-05,raccoon
2012-11-06,rabbit
...Asumiendo que tu directorio actual sea data-shell/data/,
¿qué comando usarías para producir una tabla que muestre el recuento
total de cada tipo de animal en el archivo?
grep {deer, rabbit, raccoon, deer, fox, bear} animals.txt | wc -lsort animals.txt | uniq -csort -t, -k2,2 animals.txt | uniq -ccut -d, -f 2 animals.txt | uniq -ccut -d, -f 2 animals.txt | sort | uniq -ccut -d, -f 2 animals.txt | sort | uniq -c | wc -l
La opción 5 es la respuesta correcta. Si se te dificulta entender por
qué, intenta probar los comandos, o las subsecciones de los
pipelines (sólo asegúrate de estar en el directorio
data-shell/data).
Adición de datos
Considera el archivo animals.txt, utilizado en el
ejercicio anterior. Después de estos comandos, selecciona la respuesta
que describe el contenido del archivo animalsUpd.txt:
- Las tres primeras líneas de
animals.txt - Las dos últimas líneas de
animals.txt - Las tres primeras líneas y las dos últimas líneas de
animals.txt - La segunda y tercera líneas de
animals.txt
La opción 3 es la correcta. Para que la opción 1 fuera correcta
tendríamos que haber ejecutado sólo el comando head. Para
que la opción 2 fuera correcta tendríamos que haber ejecutado sólo el
comando tail. Para que la opción 4 fuera correcta
tendríamos que haber usado un pipe para enviar la
salida de head a tail -2 de esta forma:
head -3 animals.txt | tail -2 >> animalsUpd.txt
-
catmuestra el contenido de sus entradas. -
headmuestra las primeras líneas de su entrada. -
tailmuestra las últimas líneas de su entrada. -
sortordena sus entradas. -
wccuenta líneas, palabras y caracteres en sus entradas. -
*coincide con cero o más caracteres en un nombre de archivo, por lo que*.txtcoincide con todos los archivos que terminan en.txt. -
?Coincide con un solo carácter en un nombre de archivo, así que?.txtcoincide cona.txtpero noany.txt. -
command > fileredirige la salida de un comando a un archivo. -
first | secondes un pipeline: la salida del primer comando se utiliza como entrada para el segundo. - La mejor manera de usar la terminal es utilizar pipes para combinar programas sencillos de propósito único (filtros).